
Three Key Hurdles Restraining the Growth of the Tissue Engineering Field

The interdisciplinary field of regenerative medicine – comprised of tissue engineering (TE), cell therapy (CT), and gene therapy (GT) – is growing rapidly, characterized by groundbreaking therapeutic advances that have the potential to change how healthcare providers deliver care. The fields of CT and GT have been the primary contributors to this growth in the past decade, demonstrated by the increase in available funding and marketed products, while in comparison, the field of TE has lagged behind. We believe this delayed maturation of TE is mainly due to the greater relative complexity of tissue engineered medical products (TEMPs) compared to cell and gene therapies. Generally, cell therapies transfer intact, live cells into a patient and gene therapies introduce, remove, or change a patient’s genetic code to treat disease, whereas TEMPs are engineered to repair, modify, or regenerate a patient’s cells, tissues, and organs or their structure and function, or both. This elevated complexity, which refers to both the structural composition of the end-product as well as the associated manufacturing process, has manifested itself in three key hurdles currently holding back TE:
- Complex manufacturing requirements
- Undefined regulatory pathways
- Insufficient reimbursement streams
COMPLEX MANUFACTURING REQUIREMENTS
TEMP manufacturing is relatively more complex than for cell and gene therapies due to the required raw materials, uncertainties of quality control, and difficulties scaling production capabilities.
Required Raw Materials: Compared to CT and GT, TE requires additional raw materials in the form of cells, scaffolds, and signaling molecules. It may seem that these ingredients can be combined according to a pre-determined recipe; however, the desired end-product greatly influences the quantity and type of ingredients used and raises several questions for manufacturers. Which signaling molecules induce appropriate cellular differentiation? Does the scaffold provide a suitable environment for cellular growth and vascularization? Does the end-product mimic the appropriate structural and mechanical properties? The answers to many of these questions remain largely uncertain.
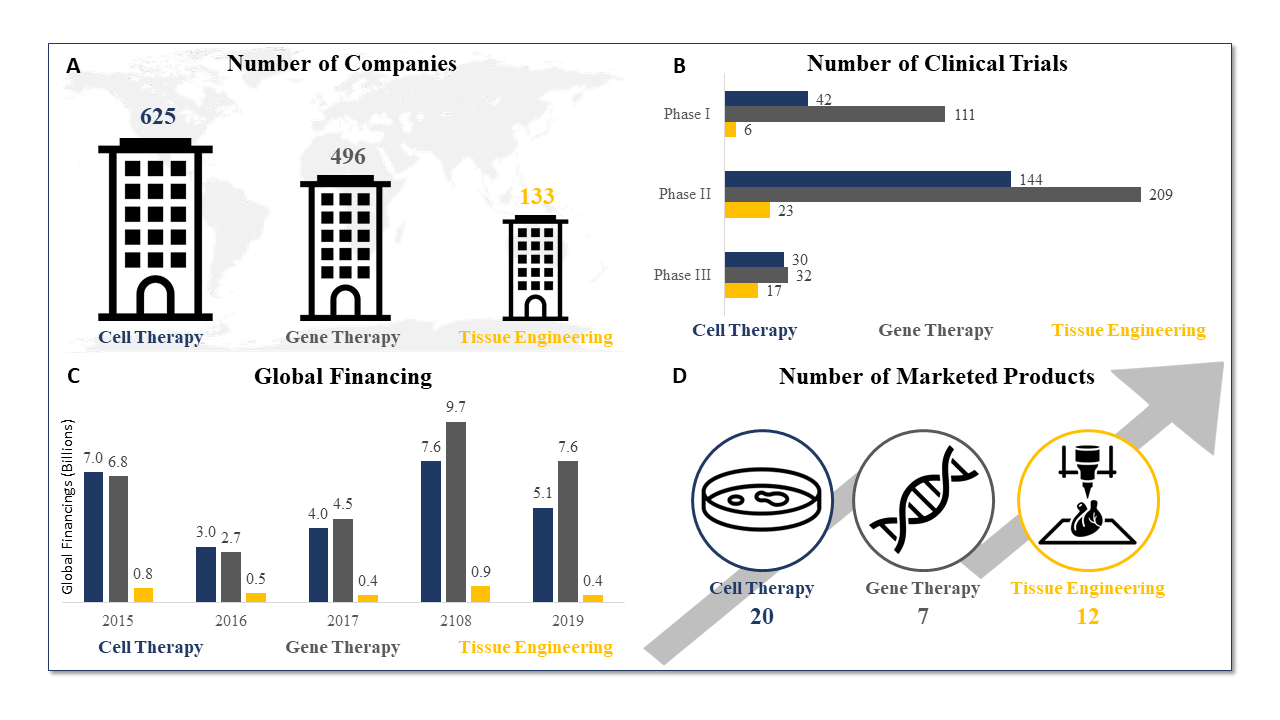
Temporal Comparison of Cell Therapy, Gene Therapy, and Tissue Engineering (Source: Data from the Alliance for Regenerative Medicine, July 2020)
Quality Control Uncertainties: Manufacturing TEMPs consistently raises quality and reproducibility issues, given it is often not feasible to only check for quality and consistency following the completion of the TEMP manufacturing process. There are also limited options for in-line non-destructive testing of critical quality attributes (CQAs) to accurately assess TEMPs prior to completion.
Difficulties Scaling Production Capabilities: TEMPs are usually manufactured by manually seeding scaffolds with cells. While this is enough for establishing proof-of-concept, manufacturers face considerable challenges when scaling for clinical production. These challenges are exacerbated when attempting to establish commercial-scale production capabilities without the infrastructure or technology required to ensure end-product quality and reproducibility. This is seen with Dermagraft®, a tissue-engineered skin substitute approved for treatment of diabetic foot ulcers and one of the most infamous TEMPs marketed. Clinical trials for Dermagraft initiated in 1991, with expectations of FDA approval by 1995. The product was not approved until 2001. Despite achieving approval and showing clinical benefit, Dermagraft has repeatedly failed to turn a profit in part due to challenges with manufacturing, regulatory, and reimbursement, showing how complicated TEMPs can be versus other therapies including cell and gene therapies.
UNDEFINED REGULATORY PATHWAYS
While regulatory pathways for TEMPs exist, they are relatively undefined compared to those for cell and gene therapies, and do not appropriately handle the added complexity of TEMPs. With such uncertainty in regulatory oversight, it is critical for TEMP investigators to align with the FDA early in development, though deciding where a product falls and how to regulate it remains unknown. Regulatory bodies make decisions based on the quality and consistency of available data but, as noted, there are limited available options for in-line, non-destructive testing of CQAs. Without empirical evidence, it becomes increasingly difficult to demonstrate necessary levels of quality and consistency. If investigators clearly understood the supporting science and could readily demonstrate appropriate levels of quality and consistency, regulatory bodies could more easily differentiate one product from another and make systematic decisions on how best to regulate TEMPs. Additional TEMP complexity is currently forcing many investigators to submit low-quality and inconsistent data. Thus, while many regulatory bodies have made considerable progress toward streamlining regulation, the baseline complexity of TEMPs continues to plague investigators and regulatory officials alike.
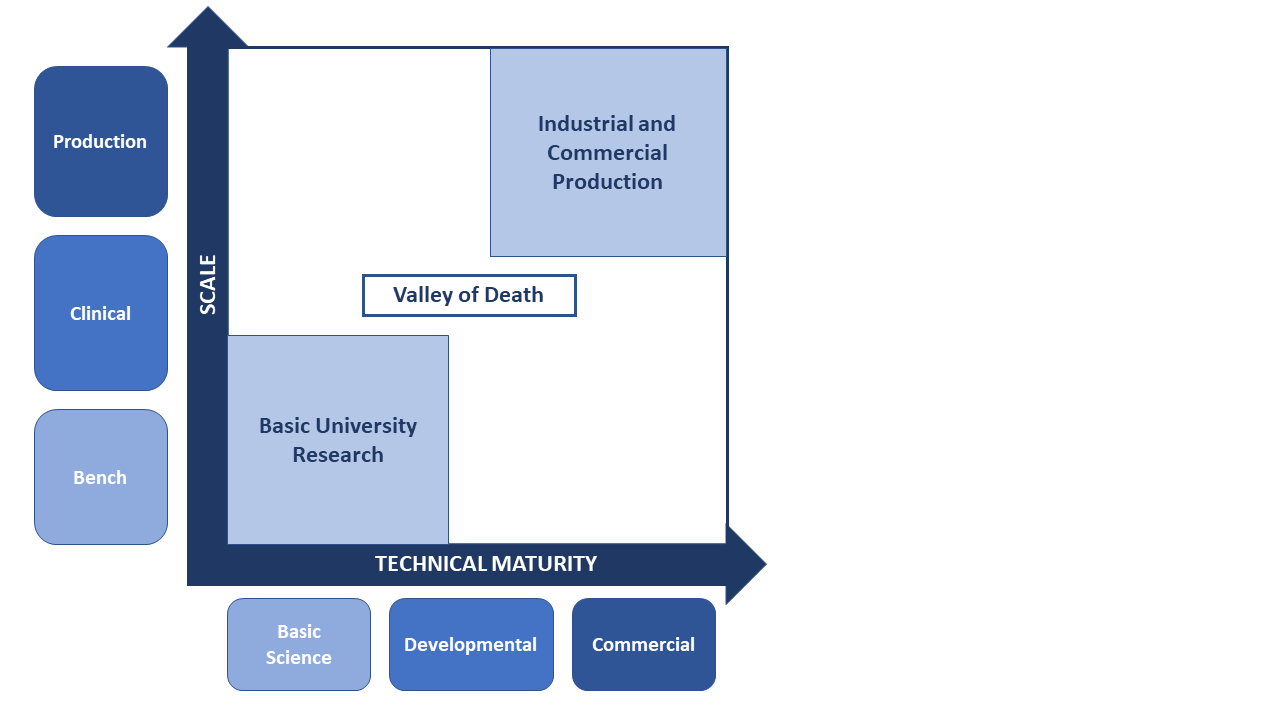
Valley of Death for TEMPs Progressing Towards Commercialization, Due to Decreased Investor Confidence (Source: CRA analysis)
INEFFECTIVE REIMBURSMENT STREAMS
Similar to the lack of industry-wide protocols for TEMP regulation, there are currently no universally accepted standards for accurately valuing and reimbursing curative cell and gene therapies and TEMPs. Curative therapies have the potential to generate long-term benefits with a single or limited number of therapeutic applications, which are valued based on their clinical impact on disease, quality of life for patients, overall healthcare system costs, and added societal value, such as increased productivity, avoided hospital visits, and long-term impact on caregivers. Despite their long-term value, curative therapies are scrutinized extensively due to their often-high upfront costs, raising serious questions about how they should be funded.
Industry stakeholders are currently focused on answering these questions on cell and gene therapies. Healthcare systems around the world are beginning to evolve their policies and reimbursement systems to accommodate curative cell and gene therapies by exploring alternative reimbursement (e.g., cost-sharing, value-based contracts) and/or financing models (e.g., reinsurance, installment payments, consumer health loans). Developing and implementing reimbursement schemes for TEMPs will be especially critical, given the high costs incurred by innovators and manufacturers in clinical development stages. Looking ahead, there is potential to apply newly developed reimbursement schemes for curative cell and gene therapies to TEMPs to help these costly and more complex products be effectively valued and reimbursed by healthcare systems.
CONCLUSION
BIBLIOGRAPHY
ASTM F2211-13, Standard Classification for Tissue Engineered Medical Products (TEMPs), ASTM International, West Conshohocken, PA, 2013, www.astm.org.
Dodson BP, Levine AD. Challenges in the translation and commercialization of cell therapies. BMC Biotechnol. 2015;15:70. Published 2015 Aug 7. doi:10.1186/s12896-015-0190-4.
Gardner, John, and Andrew Webster. 2016. “The social management of biomedical novelty: Facilitating translation in regenerative medicine.” Social Science & Medicine 156: 90-97.
Kim, Y. S., Smoak, M. M., Melchiorri, A. J., & Mikos, A. G. (2018). An Overview of the Tissue Engineering Market in the USA from 2011 to 2018. Tissue Engineering Part A. doi:10.1089/ten.tea.2018.0138.
Mount, Natalie, Stephen Ward, Panos Kefalas, and Johan Hyllner. 2015. “Cell-based therapy technology classifications and translational challenges.” Biological Sciences (The Royal Society Publishing) 307 (1680).
O’Donnell, B. T., Ives, C. J., Mohiuddin, O. A., & Bunnell, B. A. (2019). “Beyond the Present Constraints That Prevent a Wide Spread of Tissue Engineering and Regenerative Medicine Approaches.” Frontiers in Bioengineering and Biotechnology, 7. doi:10.3389/fbioe.2019.00095.
Pangarkar, Nitin, et al. “Advanced Tissue Sciences Inc.: Learning from the Past, a Case Study for Regenerative Medicine.” Regenerative Medicine, vol. 5, no. 5, 2010, pp. 823–835., doi:10.2217/rme.10.66.
Yadav, Vikramaditya, Roza Ghaemi, and Lim Siang. 2019. “Improving the Rate of Translation of Tissue Engineering Products.” Advanced Healthcare Materials 8 (19).
About the authors
The views expressed herein are the authors’ and not those of Charles River Associates (CRA) or any of the organizations with which the authors are affiliated.
Lev Gerlovin is a Vice President in the Life Sciences Practice at CRA with more than 12 years’ experience in life sciences strategy consulting, focused on commercial and market access strategies.
Andrew Thomson is a Consulting Associate within the Life Science Practice at CRA with several years of experience in commercial strategy consulting with pharmaceutical and biotech clients alike.
Jack Vailas is an Associate within the Life Sciences Practice at CRA with several years of experience with biotech innovation and commercial strategy.
Subscribe to Our Email Newsletter
Stay up-to-date on all the latest news from the 3D printing industry and receive information and offers from third party vendors.
Print Services
Upload your 3D Models and get them printed quickly and efficiently.










You May Also Like
Making 3D Printing Personal: How Faraz Faruqi Is Rethinking Digital Design at MIT CSAIL
What if your 3D printer could think more like an intelligent assistant, able to reason through a design idea, ask questions, and deliver something that works exactly the way the...
Reinventing Reindustrialization: Why NAVWAR Project Manager Spencer Koroly Invented a Made-in-America 3D Printer
It has become virtually impossible to regularly follow additive manufacturing (AM) industry news and not stumble across the term “defense industrial base” (DIB), a concept encompassing all the many diverse...
Heating Up: 3D Systems’ Scott Green Discusses 3D Printing’s Potential in the Data Center Industry
The relentless rise of NVIDIA, the steadily increasing pledges of major private and public investments in national infrastructure projects around the world, and the general cultural obsession with AI have...
Formlabs Teams Up with DMG MORI in Japan
In late June, Nick Graham, Chief Revenue Officer at Formlabs, announced on LinkedIn that the company had partnered with DMG MORI, one of the world’s leading machine tool companies, to...



















